
Harmonic Discovery
Fifty Years partners with Harmonic Discovery to design multi-target drugs


The earliest written record of medical therapeutics is the Ebers Papyrus, a 20-meter-long, 110-page medical scroll that describes how to mix herbs, shrubs, leaves, minerals, and animal poop to cure depression, diabetes – or even destroy disease-causing demons!
It's safe to assume the FDA wouldn't approve all of these ancient Egyptian treatments! However, the papyrus did contain the first ever blockbuster drug: willow bark as a general pain reliever, though its route to market was a bit circuitous. 3,500 years after the Ebers Papyrus, Reverend Edward Stone, a clergyman from Oxfordshire, dried willow bark and pounded it into a powder to cure 50 people of “aguish and intermitting disorders” (i.e., fever). It took another hundred years before chemists at Bayer purified and acetylated the active chemical in willow bark, salicylic acid, and gave it the marketing name Aspirin. With aspirin, Bayer supercharged the nascent industry of pharmaceuticals, and within two decades, aspirin was one of the most popular drugs in the world.
Soon after the success of aspirin, Bayer started pioneering synthetic and rational drug discovery methods. Chemists at Bayer realized that making simple modifications to synthetic chemical structures could produce novel therapeutics. Starting with barbital (a sleep-inducing drug) they swapped a moiety (ethyl → phenyl) to create phenobarbital, an effective treatment for epilepsy that is still used today. This sparked the era of drug discovery – a mad dash to find therapeutics by chemically modifying synthetic compounds (the ‘methyl, ethyl, propyl’ era that tested variations in structure). As our understanding of biology grew, drug design started targeting specific proteins in order to solve diseases (protein–ligand). Eventually, we created libraries of molecules and screened them for activity against key enzymes in the body (‘throw them at the wall and see what sticks’). Thirty years ago, our tools and computational ability finally allowed us to rationally design drugs based on protein structure (the first unequivocal example of a rational drug design was dorzolamide in 1995). This is the current paradigm of drug discovery – one drug for one target to cure one disease.
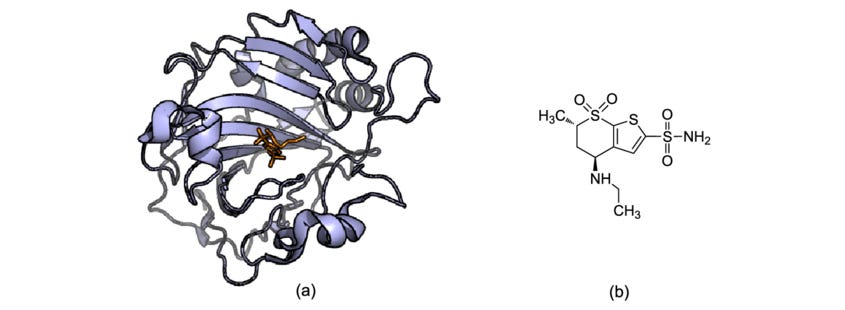
Unfortunately, the human body is a bit like 4 billion years of accumulated spaghetti code; diseases caused by just one misbehaving enzyme aren’t typical. Take cancer for example. It might take up to 10 mutations before a cell turns cancerous, and this process might dysregulate the expression of hundreds of proteins. Designing a drug to target just one misbehaving protein is tremendously difficult – there are only 812 proteins with approved therapies out of the ~20,000 in the human proteome. Taking multiple drugs to target the multiple mutations for something as complicated as cancer is dangerous – only so many drugs can be stacked before the liver becomes permanently damaged.
Kinases are the perfect example of spaghetti code. Kinases take phosphates from biology’s energy molecule, ATP, and attach them to specific amino acids like serine, tyrosine, and threonine. This is called phosphorylation. Tacking a phosphate group onto a protein causes the protein to change shape and affects how it interacts with water. Phosphorylation can also switch a protein from inactive to active or can change its function altogether. In the infinite wisdom of random walks, biology uses kinases – which turn proteins on and off – in highly conserved signaling cascades that control basic cellular functions.
Kinase signal cascades can push cells to become cancerous. The most well-studied cascade is the MAPK cascade, which begins with a stress signal at the cell surface. This phosphorylates a bunch of kinases, which in turn phosphorylate more kinases, which go on to phosphorylate a bunch of other proteins, which then leads to changes in gene expression that promote cell proliferation, survival, and migration. This is why the mutation and subsequent dysregulation of kinases in the MAPK cascade can lead directly to cancer. Take BRAF, a kinase in the MAPK signal cascade. A mutated BRAF is found in 50% of melanomas and causes a 500-fold increase in phosphorylation for 60 other proteins. This single supercharged kinase tells the cell to never stop growing and to move around the body – the definition of cancer.
This is why kinase inhibitors can be such powerful tools in the fight against cancer. Scientists understood that BRAF mutation helps cause melanomas, so they designed drugs to inhibit BRAF mutation, resulting in drugs like vemurafenib, dabrafenib, and encorafenib. Unfortunately, kinase inhibitors muck with variables deep in the spaghetti code that is biology. Kinases don’t just phosphorylate other proteins, they also form structures with other proteins that function in other signaling cascades. By inhibiting one kinase, you naturally affect the other proteins which would normally form structures with the uninhibited kinase. This is called crosstalk. Often, kinase inhibitors inadvertently target similar proteins that come earlier in the cascade, amplifying the changes to the cell’s signaling, a phenomenon known as retroactivity. Crosstalk and retroactivity lead to the off-target effects that generate a different kind of bug in our spaghetti code. These bugs have terrible consequences for patients, like cardiotoxicity, hypertension, hypothyroidism, skin reactions, proteinuria, or even more cancer (as is the case with vemurafenib and dabrafanib to treat BRAF!). We deserve more from our kinase inhibitors.
Fortunately, there is a better way, and aspirin is the clue. Aspirin is actually a very special drug – it is extremely promiscuous. Aspirin has been predicted to target over 23 proteins. It’s serendipitously polypharmacological, and because of its numerous molecular targets, it is a treatment for cardiovascular protection, fever reduction, pain relief, and may also prevent cancer (thanks Egyptians!). It’s an example of a drug that is almost impossible to design from a rational, computationally based, first-principles approach. But what if we could design kinase inhibitors that affect only the parts of cascades we care about, while modifying the surrounding cascades to avoid those terrible off-target effects?
Enter Harmonic Discovery, who are designing drugs to inhibit multiple kinases in order to cure cancer, autoimmune dysfunctions, and neurological diseases. Their drug design approach considers, from the very beginning, the multiple kinases they want to inhibit, while specifically avoiding activity with the kinases that lead to side effects. Traditional drug design attempts to target just one kinase. Trying to consider multiple kinases would multiply the complexity, so finding a kinase inhibitor for A, B, C, D, and E while avoiding activity with F, G, H, I, J, and K would be much harder (let’s say 10x harder for argument’s sake) than just finding an inhibitor for A. Harmonic Discovery fundamentally compares the structures of these kinases to create a Venn diagram – they look for motifs that exist only in the desired targets and avoid motifs that are present in the off-targets. Considering only those two circles of the Venn diagram lowers the computational complexity of finding a ligand from 10x to 2x, turning the intractable into tractable.
This is the exact insight that cofounder and CEO Rayees Rahman kept returning to during his PhD in Biophysics and System Pharmacology at the Icahn School of Medicine at Mount Sinai. He eventually realized that it is now computationally tractable to design drugs that specifically target multiple kinases and avoid off-target kinases. He pioneered the fundamental computational approach during his PhD. He then met cofounder and CSO Marcel Patek, a 25-year drug hunter veteran at Sanofi and Icagen, who immediately saw the potential to discover a new class of kinase inhibitors. Cofounder and COO, Jason Lee, who received his MD after having been a biotech banker at Deutsche Bank and investor at OrbiMed, also joined the team. Together, they’re building the next generation of computational drug discovery tools for multi-target drugs, enabling a world where we can rationally design more drugs like aspirin. With this platform, they aim to keep kinase inhibitors from creating random traffic jams and roadblocks in the cascade, so patients don’t inadvertently leave the hospital with a disease worse than the one that brought them to the hospital. Instead, Harmonic will have precision control of signals across the whole cascade, leading to extremely safe cures for currently intractable diseases.
At Fifty Years, our sweet spot is supporting founders at the earliest stages of building deep tech companies that can generate huge financial outcomes and create massive positive impact.

Deep tech: Harmonic Discovery is building the tools and approaches to support a new era of drug discovery, where one drug is designed for multiple targets. They’re starting with kinase inhibitor design in order to cure cancer.
$1b yearly revenue potential: Harmonic Discovery’s first market is the design of kinase inhibitors, which is already a >$60B yearly marketplace.
Massive positive societal impact: Just like the first chemists at Bayer ushered in the era of pharmaceutical drug discovery that gave us sedatives, antibiotics, and even aspirin (!), Harmonic Discovery will usher in a new era of drug design based on targeting multiple chemical pathways at once.
Inspired by Harmonic Discovery’s vision and execution, we at Fifty Years were proud to join its Pre-Seed round led by Innovation Endeavors and are now excited to deepen our partnership in their Series Seed.